We all have a story about 羅時 Raji דוד
-

Condolences
My name is Raji too. Some time at the dawn of the internet I was curious to see if Raji.com was available and found this site. It might have been Very late 90s or early 00s. I don’t remember anything except being very surprised that Indian Jews existed. Whenever the subject of India’s religious diversity…
-
Raji memories
I have very fond memories of Passovers at my Bubbe and Zeyda’s house (Doris and Stanley), when Lee and I wanted to hangout with Raji, Hari, and Ravi and watch them play billiards. Even though we were a lot younger, you all still humored us! The other memory I’d like to share is when Raji…
-
The Stories I Can Print
Hey man, Remember that time we had to rescue a compromised server from a chicken coop in Tom’s River New Jersey next to a garage full of combustibles? The jabronis running the operation didn’t want to let you use their toilet so they told you to go pee on a tree in the yard. And that…
-
Godzilla and お好み焼き
I’m one of the lucky ones. My wife and I got to meet Raji when we were in Tokyo in 2019. He was like bottled energy. Fun, polite, generous, and with a great sense of humor. We visited Godzilla and waited in a nice café. Then we went to an Okonomiyaki place for our first…
-

Tributes
Joshua Barry Raji became a good friend over the last few years. He was a funny guy with just enough NY moxy to make you laugh while telling you how much better you looked when you first met. We often spoke baseball and his dream was to have Ohtani join the Mets. I hope this…
-
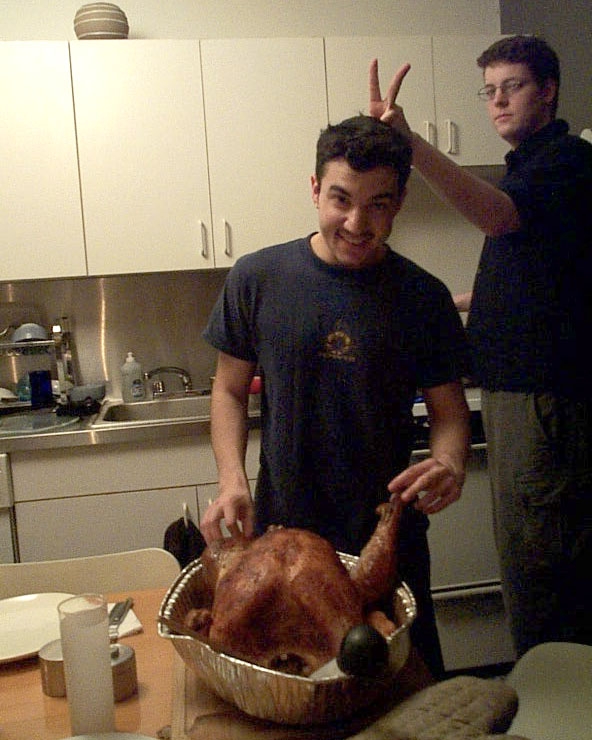
Thanksgiving with Raji
As was said over and over in the memorial service, Raji loved food–whether taking people to his favorite restaurants, cooking, or just sharing a meal. I also heard a number of people mention Thanksgiving with Raji. I still remember one of my earliest “adult” Thanksgiving dinners was at his awesome apartment in Dumbo, back when…
-

Photos of Raji in Tokyo, year 2000
In 2000, Ravi and I took a trip to Japan and Southeast Asia, touring around for about a month. We stayed in Tokyo and visited with Raji during this trip. These are just some photos I thought I would share…with some typical Raji moments…laughing hysterically, eating, riding a bike, and hanging around in Tokyo. Submitted…
-

Abridged Tokyo Hacker’s guide – by TheTokyoFixer (rajmahal)
(In March 2018, Ale and I saw Raji in Japan. I exchanged emails with him the months before, planning and also just getting giddy with anticipations. I was reading those emails, and found this.) – Don’t exchange cash – use your ATM card in the 7-11 Bank at the airport, or any 7-11, Family Mart…
-
I will miss you Raji
Everyone who has ever met Raji has a Raji story and I count myself as one of the fortunate few to have a few Raji stories. I first met Raji in a server closet at Reuters trying to hire him to come be our SE for the company. I knew in a instant I was…
-
You Will Be Missed
I met Raji when we were very young and were friends and classmates through High School. Even though we weren’t best friends, our friendship met a lot to me and I loved spending time with him. We sat next to each other in orchestra and did several after school activities together. We had so many…
-

Hi Raji-san,
Our hearts are saddened by your loss. Whenever I remember you, you are always smiling. Yes, even now and forever. The great work you did here ensures we will never forgot. Your response was quickly and we could communicate closely. You always cared about and kind to us, didn’t you? When we worked late, you…
-

started as a colleague – became a friend
Hey Raji, we met for the first time in May 2018. You had just started with us and I came to Tokyo on a business trip. Right at the start you broke the leg in your bathroom and had to hold out with crutches and painkillers. What a start. You can also see that in…
-

Hey Raji,
When i was 11, i moved to Ardsley, NY. I still clearly remember the day i met you, Raji. On my first day at Ardsley Middle School, I had no idea what to expect in the cafeteria. You found me and took me to where you were sitting with bunch of friends. For a stranger…
-

Rajaaaaaaaaaaa
“Rajar Rajar Rajar”… Missing the laugh of yours when I call you “Rajar”. You were the most honest person I’ve ever met. I always wondered how you could have such a big and warm heart. I’m glad that I’ve met you in my life. Thank you to Kana-chan ne! We miss you Rajar!! You silly!!!!!!!…
-
My friend Raji
I first met Raji through Jim Moynihan in November 2019 in Tokyo. Jim invited me to join the tennis group in Tokyo, and Raji was a key member of that group, even taking over the coordination of it when Jim left Japan in 2021. I remember that we had an instant connection for some reason.…
-
Good times in Tokyo
Hey dude. Why so fast? 🙂 I looked into my photo album and cannot believe it, but couldn’t find a photo of you in it. Why don’t I have a photo of you while I clearly remember the days I spent with you in Tokyo? One day a friend from my college time part-time job…
-

Raji and Me @ IF
I mostly know Raji through his brother Ravi, who has been one of my best friends throughout my life, and who I met in college. That said, I got to know both Raji and Hari (and their mother) over the years, and as anyone who knows the Krishaswamis can tell you, they are a tight…
-

I will miss you, my friend
I met you at Shahrod’s house in Tokyo and we became close friends. We even had friends in common back in New York and DC, including my brother-in-law Sean. You introduced me to your friends in Tokyo and Mumbai and I did the same and now we have many friends in common. In Tokyo, we…
-
NY vs. NJ
Raji is from NY. I’m from NJ. Raji is a Mets fan. I’m a Yankees fan. So it shouldn’t come as any surprise that he and I were also fierce competitors on the tennis court but good friends on and off the court. He was also a big fan of the Tennessee whiskey I am…
-

Dansu Raji
Raji, I can’t remember exactly how or where we met in Tokyo, but I am pretty sure it was in a nightclub. We became friends instantly, you were cool as heck and easygoing, funny, and always up for an adventure. Tokyo could be a really lonely place, and I always felt cool calling you for…
-
My friendship with Raji
Dear Ravi and Hari, I first met Raji in Tokyo circa Year 2016. We played tennis together. Raji was introduced to me by Clay ADLER who was the Consul General at the US Embassy/Tokyo at that time. Clay was also a member of our tennis group. Raji and myself are both big…
-

Caring and fun to be with
He has been a great help to me in my work as well as in my personal life. Especially this year, I was able to see two baseball games with him, and I was able to introduce him to my family. I am sure that I will be able to go to Meiji Jingu Stadium…
-
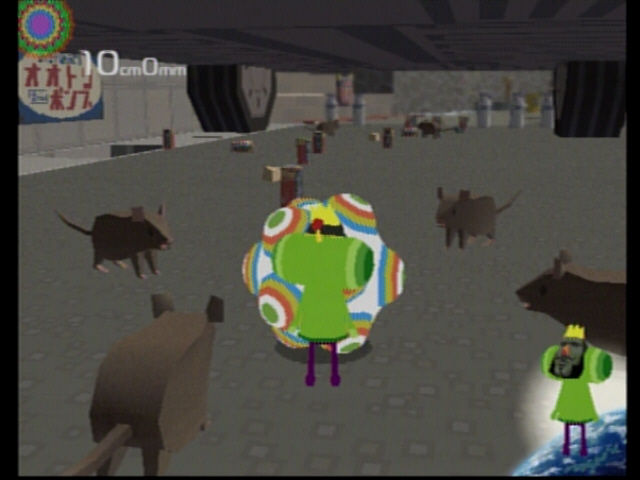
Katamari Time with Raj
So many stories with Raj, so I have to start somewhere. Wikipedia tells me it had to be early Fall 2004.. Raji had just nabbed a copy of Katamari Damacy and I was headed to his apartment with a 6-pack of Grolsch. I’d read about the game and was looking forward to tracking it down.…
-

The Nomad
By Nick Olsen The Nomad The nomad, the foraging soul, the seasonal traveler and trader merchant represents who we are in our life path. The nomad brings a special set of skills to not only thrive in their market speaking local languages understanding regional customs. The nomad that exemplifies these special qualities bring a tale…
-

Murakami hits 2 homers tonight
Hey, Raj. One of your favorite Swallows player Murakami hit two homers today. Raj, you and I drank beer in Tokyo, the US, Holland, and many other places, but I never thought you started beer at TGI Friday’s in heaven now. I haven’t shown you the picture of you dressed up as a cop for…
-
スワローズ万歳
ラジ 明日侍ジャパン観に行くよ! 村神様のホームラン願って下さい! 1回でもいいからラジとスタジアム行きたかったな! 何回も誘ってくれてありがとうね! 明日は全力で応援してくるよ! ラジも上から見守ってて下さい👍 Submitted by nabe.
-

For Raj
It’s really only hitting me now just how long I go back with Raji and while I don’t think anyone would say that the two of us were super tight friends, we really were good friends, and got to that point even independent of the fact that he is the youngest brother of my best…
-

the one and only
親愛なるラジ 私にとってあなたは爆弾みたいな人だったよ とても刺激的な爆弾 笑って泣いて怒ってら、 性格も、仕事もプライベートも とにかく忙しい人だから なんでそんなに急ぐのかって聞くと 僕はニューヨーカーだからって言ってたね。 貴方が全力で毎日無駄なく人生を楽しんでいた証拠 わたしが、お腹空くと、ハングリーアングリーってバカにしながらリュックから大好きなワッパーバーガー出してくれるの 私のドラえもんだった 優しいラジ− 温かいラジ− 意地悪なラジ− わがままなラジ− 全て大好きだよ 最後も喧嘩してバイバイしちゃったけど ごめんね ラジが正しかった 本当に貴方は数え切れないくらい程の多くの友達に愛されてる 私も含めて 悔しいからこっちの世界でラジの分まで全力で楽しから!! ラジはゆっくり休んでね、もう急がなくていい 待っててね親愛なるラジ 大好きだよ Submitted by Kana Yamada.
-

Raji’s magic
There was a time when I worked very closely with him, though for a short period of time. I cannot help but be surprised at this sudden sad news. I have many memories of him, but I will never forget the days when we hit it off to go to “Yufuin”, a famous hot spring…
-
How we met & the broken TV
How we met: I met Raji for the first time at a small hole in the wall Japanese pub back around 2015-16. At the time, I was living in a residential suburb of Tokyo. This pub was the only place open past 10pm in this quiet neighborhood. The onwer, a Chinese lady, told me there…